Locations:

Sudden-onset abdominal pain prompts urgent visit
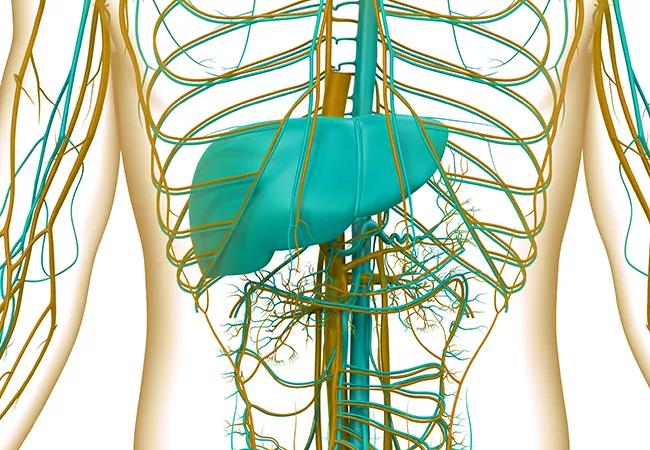
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/292e523e-f98d-4632-b4b9-aae9a879e79d/21-CNR-2572060-CQD-650x450-1_jpg)
Human Internal Digestive Organ Liver Anatomy
A 42-year-old woman with sudden-onset abdominal pain presented to Cleveland Clinic in need of an urgent evaluation. She had been diagnosed with paroxysmal nocturnal haemoglobinuria (PNH) five years earlier and had missed multiple follow-up appointments. Despite having a large PNH clone (>50% of PNH granulocytes), the patient had been asymptomatic with only mildly elevated haemolytic parameters; treatment with eculizumab had been deferred.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
On examination, the patient was afebrile and her vital signs were stable, explains Jaroslaw P. Maciejewski, MD, PhD, Chairman of the Department of Translational Hematology and Oncology Research at Taussig Cancer Institute. Slight scleral icterus was noted. Her liver was palpable with soft edges, and she exhibited right upper-quadrant tenderness without peritoneal signs. The spleen was also palpable approximately 3 cm below the left costal margin.
Laboratory tests revealed elevated liver enzymes: aspartate aminotransferase 164 U/L (normal 13–35), alanine aminotransferase 116 U/L (normal 7–38), total bilirubin concentration 2·9 mg/dL (normal 0·2–1·3). D-dimer concentration was raised at 11 420 ng/mL (normal <500), lactate dehydrogenase was 1 111 U/L (normal 135–214), and her haemoglobin concentration was low at 9·2 g/dL (normal 11·5–15·5).
A CT scan and MRI of the patient’s abdomen showed splenomegaly with a heterogeneous appearance of the liver with hypointensity along the vascular territory, tapering occlusion of the hepatic veins, and focal narrowing at the level of the confluence. These findings were
consistent with chronic Budd–Chiari syndrome, a life-threatening condition in which the hepatic veins are blocked or narrowed by a clot.
After consultation with the hepatology service, no acute interventions were recommended. The patient was started on apixaban 5 mg twice a day and the anticomplement factor 5 inhibitor, ravulizumab — beginning with a loading dose of 3000 mg intravenously (weight-based
dose), followed 2 weeks later with a maintenance dose of 3600 mg every 8 weeks. Notably, deep sequencing of the PIGA gene showed five mutations: two frameshift (p.F150fs, p.E120fs), two missense (p.C81Y, p.S155Y), and one splice site (c.981+1G>A) at a mean total variant allele frequency of 16%.
Advertisement
The patient showed significant clinical improvement with resolution of her abdominal pain after 2 weeks of treatment.
PNH is a progressive, non-malignant disorder genetically defined by somatic loss-of-function mutations of the PIGA gene in haematopoietic stem cells. The mutations result in impaired biosynthesis of glycosylphosphatidylinositol (GPI), a glycolipid structure that attaches specific proteins to the cell membrane. Impaired GPI-anchoring results in a deficiency of all GPI-linked proteins on the membranes of blood cells derived from the mutant clone. Among the GPI-linked proteins affected, deficiency of CD55 and CD59 on erythrocytes is responsible for increased sensitivity to complement-mediated intravascular haemolysis—the cardinal symptom of PNH.
In addition to haemolysis, PNH causes bone marrow failure and thrombophilia—the most common cause of death. Although thrombosis can occur at any site, unusual locations are typical of PNH-related hypercoagulability, including hepatic vein thrombosis or Budd–Chiari syndrome (also called hepatic vein thrombosis), which is seen in 10%–25% of patients.
Thrombosis can be either the initial presentation or can occur in the course of the disease; it is likely to be related to haemolysis as shown by the preventive effects of complement blockade treatment on the rate of this complication.
This case was initially outlined in the Lancet and has been abbreviated for ConsultQD.
Advertisement
Advertisement

First-of-its-kind research investigates the viability of standard screening to reduce the burden of late-stage cancer diagnoses

Global R&D efforts expanding first-line and relapse therapy options for patients
Study demonstrates ability to reduce patients’ reliance on phlebotomies to stabilize hematocrit levels

A case study on the value of access to novel therapies through clinical trials

Findings highlight an association between obesity and an increased incidence of moderate-severe disease
Cleveland Clinic Cancer Institute takes multi-faceted approach to increasing clinical trial access 23456
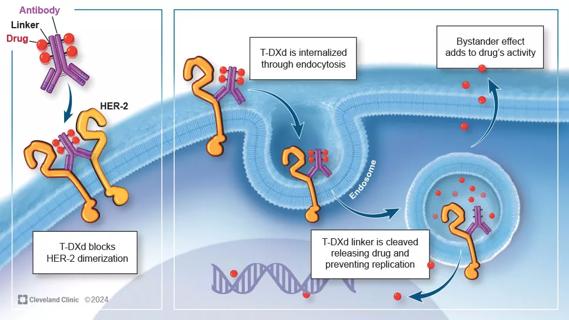
Key learnings from DESTINY trials

Overall survival in patients treated since 2008 is nearly 20% higher than in earlier patients